
IN-SILICO ANALYSIS OF THE STRUCTURE AND BINDING SITE FEATURES OF THE 3CL PROTEASE FROM SARS-COV-2: PARAMETERIZATION FOR VIRTUAL SCREENING PROTOCOLS
Maria Eduarda Alves Esteves, Tácio Vinício Amorim Fernandes and Manuela Leal da Silva
The new SARS-CoV-2 virus (severe acute respiratory syndrome coronavirus 2) emerged at the end of 2019 as a global emergency. Due to its high rate of transmission and the absence of specific treatment or vaccine, around 1 million people over the world have died, according to World Health Organization until October 2020. Nowadays, thousands of people still get infected every day and many of them do not survive due to the complications of the disease associated with the acute respiratory syndrome. Thus, once the pharmacological therapy has shown to be deficient because of its non-specificity, this work intends to conduct an in silico research for possible drugs and bioactive substances, including those belonging to Brazilian biodiversity, that can act as inhibitors of the main viral protease (3CLpro) for the treatment of COVID-19.
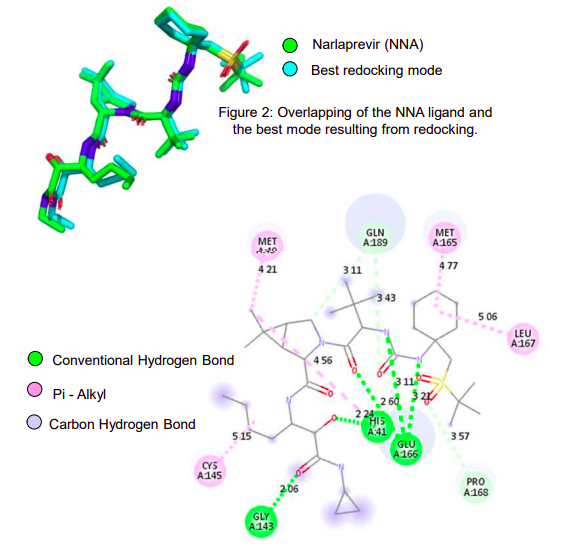
In this work, the prediction of the amino acid residues’ pKa of the receptor protein (PDBid: 6XQT) through the PDB2PQR server and the selection of the ionizable residues’ protonation probable state of the 3CLpro three-dimensional structure using the pdb2gmx module were performed as parameterization methods. The anchorage site of the ligands was delimited by the grid center x, y, z: -11, 1, 45 and size x, y, z: 32, 35, 33, respectively, involving the catalytic dyad His41 and Cys145. In the redocking stage, the exhaustiveness of 8, 16, 32, 64 and 100 were tested, with the result of less exhaustiveness being selected with the affinity calculated by Autodock Vina equal to -10.4 kcal/mol. In this step it was possible to obtain an RMSD (Root Mean Square deviation) of 0.97 Å between the original ligand of the crystal and the first model generated from the docking. It was possible to stipulate through the performed methodology the parameters for the next stage of virtual screening, whose results are under analysis.